Home

FusionInspector is a component of the Trinity Cancer Transcriptome Analysis Toolkit (CTAT). FusionInspector assists in fusion transcript discovery by performing a supervised analysis of fusion predictions, attempting to recover and re-score evidence for such predictions. As of July, 2017, FusionInspector has been included as a component of the STAR-Fusion suite.
Given a list of candidate fusion genes (as derived from running any fusion transcript prediction tool, such as Prada, FusionCatcher, SoapFuse, TophatFusion, DISCASM/GMAP-Fusion, STAR-Fusion, or other), FusionInspector extracts the genomic regions for the fusion partners and constructs mini-fusion-contigs containing the pairs of genes in their proposed fused orientation. The original reads are aligned to these candidate fusion contigs; fusion-supporting reads that would normally align as discordant pairs or split reads should align as concordant 'normal' reads in this fusion-gene context. Those reads supporting each fusion (spanning fragments and fusion-breakpoint-containing reads) are identified, reported, and scored accordingly. An overview of the FusionInspector process is shown below.
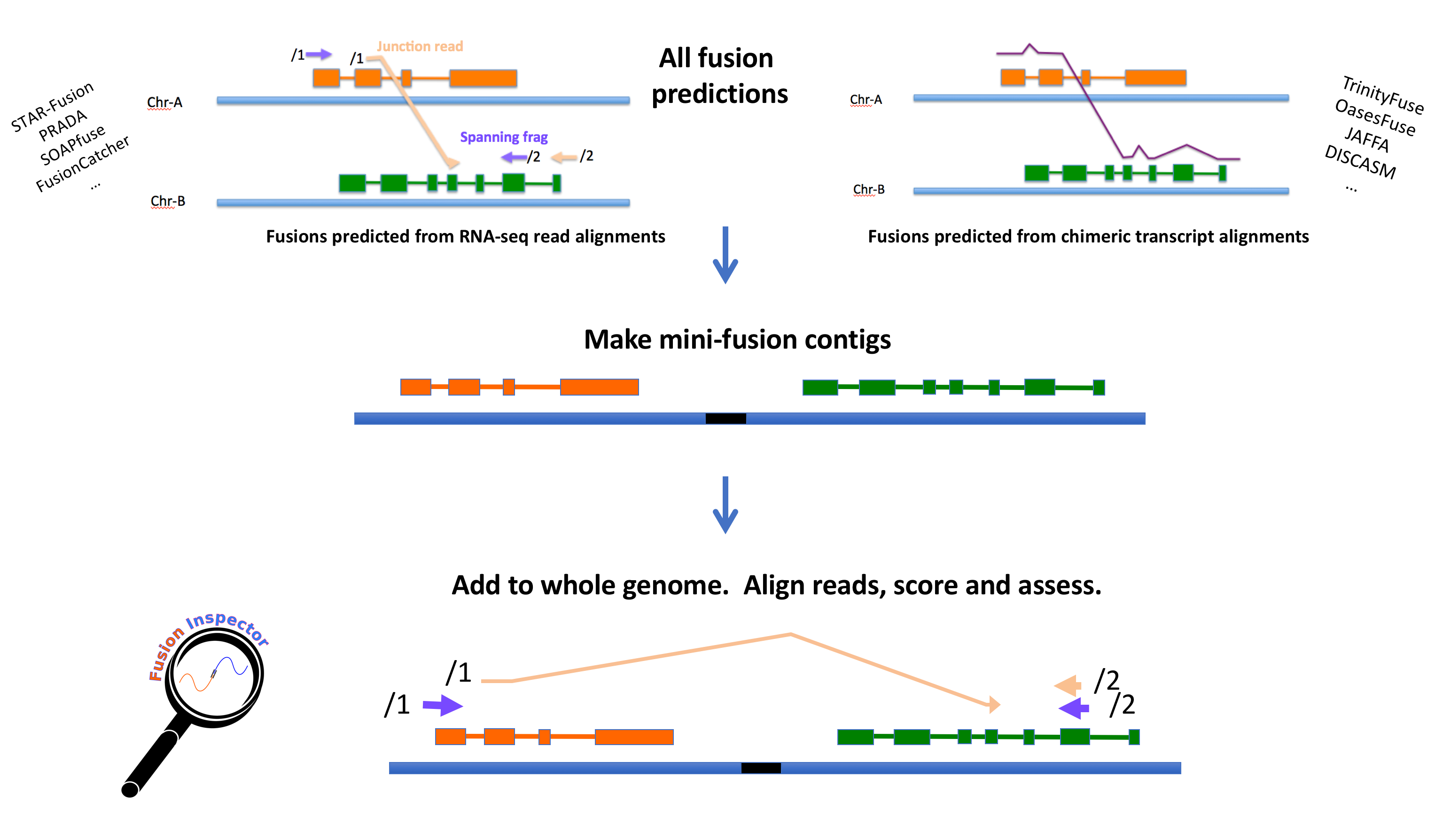
Optionally, Trinity de novo transcriptome assembly can be executed as part of the FusionInspector routine in order to de novo reconstruct fusion transcripts from the mapped reads.
The evidence for fusions as evaluated by FusionInspector are easily viewed and navigated via html-based fusion reports included as output. Alternatively, outputs in standard formats (bed, gtf, fasta) can be visualized in a genome browser such as IGV. Examples are provided below.
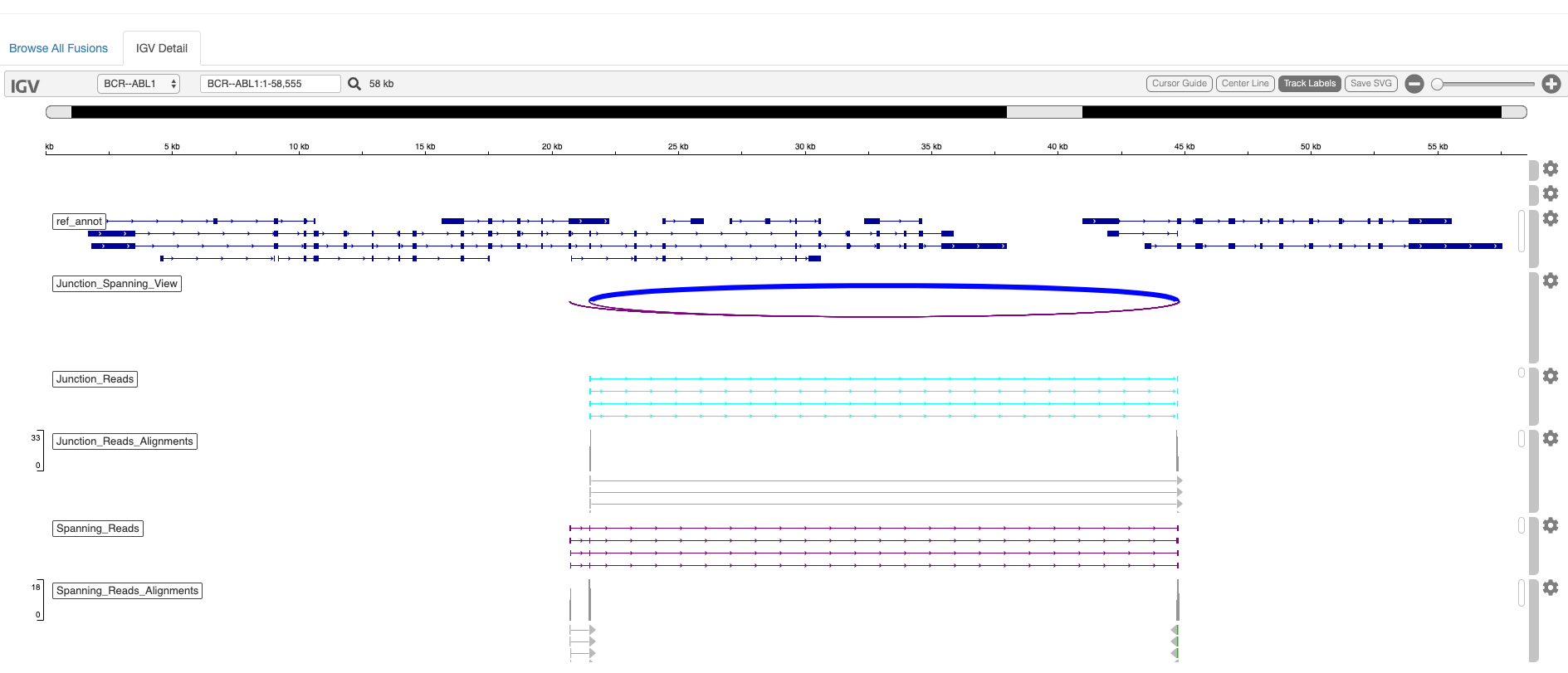
See FusionInspector Visualizations for detailed options.
FusionInspector computes metrics associated with the expression of the fusion transcript, the expression of the unfused partner genes, and examines sequence characteristics of the splice sites and positions of microhomologies observed between the partner genes. These data and related visualizations allow for reasoning about the quality of the evidence supporting the fusion transcript and whether or not the fusions likely reflect biologically relevant transcripts or instead more likely to be derived from experimental or bioinformatic artifacts.
Known cancer-relevant fusion transcripts (ie. those annotated in the COSMIC fusion database) tend to be enriched for certain properties such as being highly expressed, having breakpoints that coincide with reference gene annotation splice junctions, having few microhomologies detected between the fusion partners or near the putative fusion transcript breakpoints, and having relatively high fusion allelic ratio for the 3' fusion partner as compared to the 5' partner. Based on these features, FusionInspector can make predictions whether target fusion predictions appear to be COSMIC-like or if they have features that suggest being artifact-derived.
See COSMIC-like or Artifact-like fusion prediction analysis here.
Installing FusionInspector should be a breeze. It does require additional popular software such as the STAR aligner and samtools, but FusionInspector is written in Python and doesn't require any compilation. If you can use Docker, we have a FusionInspector Docker image and Singularity image that comes with all companion software integrated. See Installing FusionInspector for all installation details.
FusionInspector requires one or more lists of fusion candidates, with each formatted like so, as geneA--geneB:
B3GNT1--NPSR1
ZNF709--DYRK1A
ZNF844--NCBP2
RBX1--HAPLN2
FAM180B--TRIM60
CASP9--ADCYAP1
HS3ST3A1--C1QTNF2
OPTC--AP000347.4
GRIA2--ZW10
We'll call the file containing this list 'fusions.listA.txt'. Let's assume we have another such list from another source, and we'll call it 'fusions.listB.txt'.
It's ok to have a tab-delimited file containing other attributes (such as the raw output from some fusion-prediction tool) as long as the first column fits the above format.
Given this list of fusions, we'll run FusionInspector like so:
FusionInspector --fusions fusions.listA.txt,fusions.listB.txt \
--genome_lib /path/to/CTAT_genome_lib \
--left_fq rnaseq_1.fq --right_fq rnaseq_2.fq \
--out_dir my_FusionInspector_outdir \
--out_prefix finspector \
--vis
The FusionInspector Outputs include several outputs that are fully described at the link provided. If Trinity de novo assembly is included, the reconstructed fusion transcript sequences and fusion/genome alignments are provided and integrated into the visualizations.
See the 'test/' subdirectory and examine the README.txt file included. Example data and command execution info are provided.
Contact us on our google group https://groups.google.com/forum/#!forum/trinity_ctat_users
FusionInspector is primarily a collaboration between Brian Haas (Broad Institute) and Alex Dobin (Cold Spring Harbor Laboratory), and developed as part of the Trinity Cancer Transcriptome Analysis Toolkit. The igv-reports based fusion report derives from a collaboration with James Robinson.
FusionInspector is supported as part of the Trinity CTAT Project, funded by the National Cancer Institute Informatics Technology for Cancer Research